
Nacuity is the leader in innovative treatments for oxidative stress, the widely known, unaddressed disease mechanism hiding in plain sight. Oxidative stress has been implicated as a driver of blinding eye diseases and a broad spectrum of serious chronic conditions. The company is developing targeted, prescription antioxidant therapies that aim to stop chemically aggressive oxygen molecules from damaging tissue. Nacuity’s molecules are differentiated, prescription-quality, GMP-grade moieties, which, combined with innovative delivery technologies, are poised to address diseases and conditions involving oxidative stress including retinitis pigmentosa, cataract and cystinosis as well as rare neurological diseases.

The Amide Advantage
Nacuity’s refined, prescription-quality amide, NPI-001, has consequential properties for central nervous system use. NPI-001 is lipophilic and easily permeates cell membranes. In preclinical studies, NPI-001 has exhibited the ability to cross blood-brain and blood-retinal barriers.
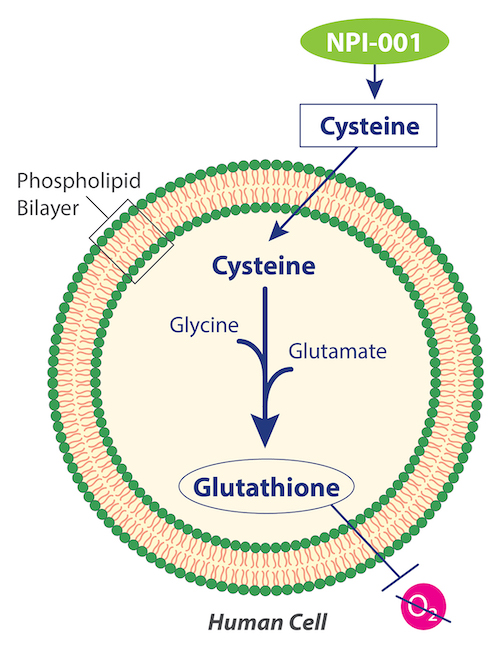
Nacuity is studying the broad clinical potential of a differentiated, purified, prescription-quality, GMP-grade N-acetylcysteine amide (NPI-001). The company’s lead candidate (NPI-001), manufactured using a patented process, is the amide form of N-acetyl-L-cysteine (NAC).
Glutathione
NPI-001 targets cells’ lack of available cysteine, which is required for intracellular biosynthesis of glutathione, the body’s most effective endogenous antioxidant. Though it is found in every cell, glutathione cannot penetrate cell walls, thus it cannot be externally supplemented, and it must be biosynthesized inside the cell. The substrates, glutamate and glycine, are often present in cells, but some cells lack sufficient cysteine to make additional glutathione.
NAC
Previous research with NAC demonstrated its mechanism of donating cysteine to boost glutathione to treat disease, replenishing glutathione within cells that are undergoing oxidative stress. NAC is approved by numerous regulatory agencies for the treatment of hepatotoxicity caused by acetaminophen overdose, involving replenishment of glutathione in the liver. It is also approved as a mucolytic agent.
NPI-001’s Advantage over NAC
Nacuity’s refined, prescription-quality amide, NPI-001 (NACA), has significant advantages over NAC. NPI-001 is more lipophilic and more easily permeates cell membranes than NAC. In preclinical studies, NPI-001 has exhibited the ability to cross blood-brain1 and blood-retinal2 barriers.
NPI-001 has the potential to treat a broad set of diseases and conditions associated with oxidative stress and reduced glutathione levels – such as slowing or stopping disease progression in retinitis pigmentosa patients with functional cones, regardless of their disease-causing genetic mutation.
Ocular Diseases Involving Oxidative Stress
Nacuity’s antioxidants, if demonstrated safe and effective, have the potential to treat several ocular diseases and conditions associated with oxidative stress and reduced glutathione levels.
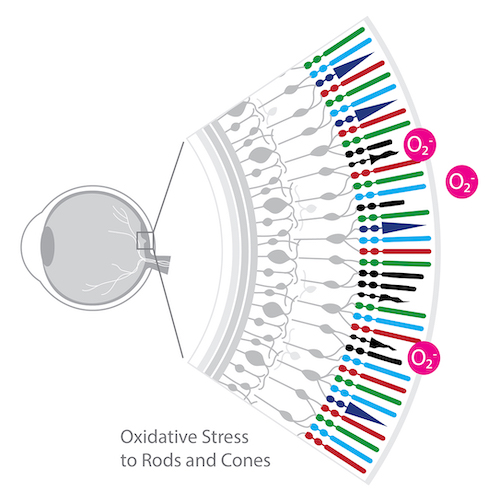
Retinitis Pigmentosa:
The extensive research of Dr. Peter Campochiaro, George S. & Dolores Doré Eccles Professor of Ophthalmology & Neuroscience, Wilmer Eye Institute, The Johns Hopkins School of Medicine involving oxidative stress in the retina provided impetus for exploring small molecule antioxidant therapies. Accumulating experimental evidence points to oxidative stress as a pathogenic factor in RP.3, 4, 5, 6, 7, 8
- An immunohistochemical study in a RP pig model suggested that the death of rods results in decreased oxygen consumption and hyperoxia in the outer retina resulting in gradual cone cell death from oxidative damage.3
- In a study using the rd1 mouse RP model, a mixture of antioxidants including alpha-tocopherol, ascorbic acid, Mn(III)tetrakis(4-benzoic acid)porphyrin and alpha-lipoic acid improved biomarkers of oxidative stress (protein carbonyl adducts and acrolein staining) and partially preserved cone function.4
- Antioxidant-treated rd10+/+ mice showed preservation of cone function, including a significant increase in photopic ERG b-wave amplitudes. 5
Mutations that cause RP initially lead to rod cell death. After rod photoreceptors die, cone photoreceptors gradually die.9
In a study of RP patients, Campochiaro et al.10 reported a significant reduction in the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) in aqueous humor and a significant increase in aqueous protein carbonyl content, compared to control subjects. In contrast, there was no significant decrease in the serum GSH/GSSG ratio or increase in carbonyl content of serum proteins. These data suggest that patients with RP may exhibit ocular oxidative stress, ultimately leading to cone death. Potent antioxidants, therefore, should promote cone survival and function in patients with RP and aqueous GSH/GSSG ratio and protein carbonyl content may provide useful biomarkers.
Cataract:
Several nonclinical studies have demonstrated the anticataract activity of N-acetylcysteine amide (NACA) in various in vivo cataract models.
Carey et al.11 have shown the efficacy of NACA against L-buthionine-(S,R) sulfoximine (BSO, a glutathione synthesis inhibitor)- induced cataract in wistar rat pups. Also, NACA was shown to inhibit dexamethasone-induced cataract formation by limiting lipid peroxidation and increasing the ratio of GSH/GSSG in rat lens.12 In addition, NACA prevented acetaminophen-induced cataractogenesis in rats.13 Furthermore, NACA decreased severity of selenite-induced cataract in rat pups.14
Nacuity is developing NPI-002 (diNACA), an analog to NPI-001, as an anticataract agent. NPI-002 has demonstrated activity against peroxide- and glucose oxidase-induced cataracts in isolated rat and pig lenses. 15
Age-related macular degeneration:
Age-related macular degeneration, commonly referred to as AMD, is a retinal degenerative disease that causes a progressive loss of central vision. AMD is the most common cause of blindness in individuals over the age of 55 years in developed countries. More than 10 million people in the United States have AMD. 16
The Age-Related Eye Disease Study (AREDS), a landmark investigation conducted by the National Eye Institute, determined that specific antioxidant supplementation can slow the progression of AMD. The AREDS2 formulation is an over-the-counter antioxidant supplement recommended for people who are at risk of developing more advanced forms of dry or wet AMD. The AREDS2 formulation contains the following nutrients: 500 milligrams (mg) of vitamin C; 400 international units of vitamin E; 80 mg zinc as zinc oxide; 2 mg copper as cupric oxide; and 10 mg lutein and 2 mg zeaxanthin.
Often diagnosed in childhood or adolescence, Stargardt disease is an inherited form of macular degeneration causing central vision loss. The condition is sometimes referred to as juvenile or early onset macular degeneration.16
- NPI-001:
Schimel et al. (2011)17 independently demonstrated that N-acetylcysteine amide (NACA) remarkably protected retinal pigmented epithelium (RPE) and photoreceptors against oxidative cell damage and death, in vitro (ARPE-19 cell culture treated with tBHP) and in vivo (light damage mouse model). They concluded that NACA may be a novel treatment in rescuing retinal function and preventing vision loss secondary to retinal degenerative diseases including AMD.
- NPI-001 and NPI-002:
A recent study18 was conducted to discern relative antioxidant effects of Nacuity’s antioxidants compared to NAC in various retinal cell cultures. This study revealed that NPI-001 (NACA), NPI-002 (diNACA) and NAC exhibited varying degrees of antioxidant activity, i.e., protected cultured rat retinal cells from a variety of stressors which were designed to mimic aspects of the pathology of different retinal diseases. A general rank order of activity was observed: NPI-001 ≥ NPI-002 > NAC. Studies are planned to further evaluate NPI-001 and NPI-002 as potential treatments for AMD and Stargardt disease.
Non-Ocular Diseases Involving Oxidative Stress
Several antioxidants like N-acetylcysteine (NAC), lipoic acid, tocopherol, probucol, and Coenzyme QlO have been investigated for their neuroprotective efficacy. Unfortunately, these agents do not cross blood-brain barrier (BBB). BBB permeability is a requirement for any neuroprotective antioxidant. Based on its ability to cross the BBB, N-acetylcysteine amide (NACA) has been evaluated for its neuroprotective effect using cell lines and animal models of neurological diseases/disorders.
Nephropathic cystinosis:
Oxidative stress plays a role in the pathogenesis of cystinosis.19 , 20
Nacuity’s small molecule antioxidants lack cytotoxicity and increase viability of human cystinotic fibroblast cells in culture. NPI-001 depleted cystine from human cystinosis fibroblast cells in culture statistically significantly greater than cysteamine.21
Heriditary cystatin C amyloid angiopathy (HCCAA):
Hereditary cystatin C amyloid angiopathy (HCCAA) is a rare dominantly inherited disease caused by a leucine to glutamine variant of human cystatin C (hCC). L68Q-hCC forms amyloid deposits in brain arteries associated with micro-infarcts, leading ultimately to paralysis, dementia and death in young adults. To evaluate the ability of molecules to interfere with aggregation of hCC while informing about cellular toxicity, cells were generated that produce and secrete WT and L68Q-hCC producing high-molecular weight complexes formed from the mutant protein. Incubations of either lysate or supernatant containing L68Q-hCC with reducing agents glutathione or N-acetylcysteine (NAC) break oligomers into monomers. Six L68QhCC human carriers taking oral NAC had skin biopsies obtained to determine if hCC deposits were reduced following NAC treatment. Remarkably, ~50–90% reduction of L68Q-hCC staining was observed in five of the treated carriers suggesting that L68Q-hCC is a clinical target for reducing agents.
Reducing agents similar to NAC, NAC-amide and NAC-methyl ester, were evaluated for their effects on oligomerization/dimerization of secreted hCC in vitro. NAC-amide and NAC-methyl ester exhibited approximately 10-fold greater activity compared to NAC.22 Based on the favorable bioavailability of the amide, N-acetylcysteine amide (NACA), over the acid, NAC,23 and likely its methyl ester,24 the pursuit of NACA therapy for treatment of HCCAA is warranted.
Gain-of-function mutation in ACOX1, Mitchell Syndrome:
Gain-of-function mutation in peroxisomal acyl-CoA oxidase 1 (ACOX1) is a very rare heterozygous missense variant (p. N237S) disorder characterized by an increase in reactive oxygen species (ROS), chronic loss of axons and progressive loss of nervous system function in pediatric patients. Patients with ACOX1 (p.N237S) exhibit loss of glia, Schwann cells and neurons. The equivalent mutation expressed in fruit flies (Drosophila, da>dACOX1N250S) also exhibited elevated levels of ROS and increased lethality. Feeding mutant flies the antioxidant, N-acetylcysteine amide (NACA), suppressed the neurodegeneration caused by gain-of-function mutation in ACOX1. Similarly, the same mutation in rodents also causes increased ROS in Schwann cells and leads to death of primary Schwann cells in culture. The effects of the mutation are reduced by treatment with NACA, resulting in increased survival of Schwann cells.25
A nerve biopsy of the first known patient with the ACOX1N237S mutation revealed a loss of Schwann cells. Similarly, overexpression of hACOX1N237S in cultured rat Schwann cells induced apoptosis, and cell death was suppressed by NACA. Given that NACA was able to counteract the effects of the ACOX1 gain-of-function mutation, antioxidant treatment was considered for patient#1 during a severe disease flare. Since NACA is not approved for use in humans, a related drug, N-acetylcysteine (NAC), was used. During a several-month course of NAC treatment, patient#1 had a dramatic improvement in peripheral nerve function. Possibly because NAC does not cross the blood-brain barrier well enough, the patient eventually succumbed to a novel and rapid onset of CNS disease. In honor of patient#1, the investigators named this novel ACOX1-mediated disease “Mitchell Syndrome” (distinct from “Mitchell's disease”, Erythromelalgia).26 Based on these findings, the pursuit of NACA therapy for treatment of Mitchell Syndrome, is warranted.





















